
| Home |
| News 02/09/2544 |
| The 'Generic-ization' of Drugs: Will Patients Benefit? |
| A Comparison of Simvastatin and Atorvastatin up to Maximal Recommended Doses in a Large Multicenter Randomized Clinical Trial |
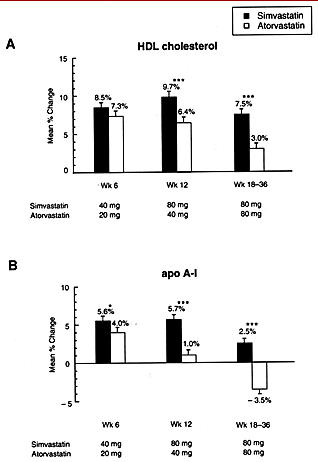
Figure 1. Least-squares mean ( ±SE) percentage change from baseline in (A) high-density lipoprotein cholesterol (HDL cholesterol) and (B) apolipoprotein A-I (Apo A-I). Asterisks indicate a significant between-group difference (*p < 0.050, ***p < 0.001).Treatment with atorvastatin led to greater reductions in LDL cholesterol and triglyceride levels compared with simvastatin for the three dose comparisons, although the magnitudes of the differences were small relative to the substantial total reductions produced by both drugs in plasma lipids and lipoproteins. Based on the average change from baseline at Weeks 6, 12 and 18 to 36, dose comparisons of simvastatin 40 mg versus atorvastatin 20 mg (Period 1), simvastatin 80 mg versus atorvastatin 40 mg (Period 2), and simvastatin 80 mg versus atorvastatin 80 mg (Period 3), produced mean reductions in LDL cholesterol of 42.4 versus 46.1%, 48.8 versus 51.3%, and 48.1 versus 53.6%, respectively ( p
Abbreviations: apo A-I, apolipoprotein A-I; HDL, high-density lipoprotein; LDL, low-density lipoprotein; LDL/HDL, ratio of LDL cholesterol and HDL cholesterol; TC/HDL, ratio of total cholesterol and HDL cholesterol
Characteristic Simvastatin N = 405) Atorvastatin (N = 408) Gender:
female 172 (42.5%) 211 (51.7%) male 233 (57.5%) 197 (48.3%) Mean (SD) baseline lipid levels (mg/dl)*:
HDL cholesterol 50 (11) 51 (12) apo A-1 150 (25) 154 (28) LDL cholesterol 209 (51) 206 (49) total cholesterol 295 (50) 292 (51) triglycerides 179 (73) 177 (67) LDL/HDL 4.4 (1.6) 4.2 (1.3) TC/HDL 6.2 (1.8) 6.0 (1.5)
*Multiplication factors for converting lipid values from mg/dl to mmol/l: HDL cholesterol and LDL cholesterol, 0.026; triglycerides, 0.011
AE: Adverse experience considered to be possibly, probably or definitely drug-related; S: simvastatin; A: atorvastatin
Study period Period 1 Period 2 Period 3 Duration 6 Weeks 6 Weeks 24 Weeks Treatment group S 40 mg A 20 mg S 80 mg A 40 mg S 80 mg A 80 mg Clinical AE 3/414 (0.7%) 3/412 (0.7%) 24/399 (6.0%) 36/404 (8.9%) 46/385 (11.9%) 92/394* (23.4%) Discontinuation due to clinical AE 0/414 (0%) 0/412 (0%) 7/399 (1.8%) 5/404 (1.2%) 9/385 (2.3%) 11/394 (2.8%) Laboratory AE 3/414 (0.7%) 3/412 (0.7%) 3/399 (0.8%) 4/404 (1.0%) 15/385 (3.9%) 48/394* (12.2%) Discontinuation due to laboratory AE 0/414 (0%) 0/412 (0%) 0/399 (0%) 0/404 (0%) 3/385 (0.8%) 16/394 (4.1%)
*Significant between treatment-group difference ( p < 0.001)
ALT = alanine aminotransferase; AST = aspartate aminotransferase; ALP = alkaline phosphatase; GGT = gamma glutamyl transpeptidase
Age/sex ALT (u/l) (ULN 25) Repeat ALT (u/l) AST (u/l) (ULN 22) ALP (u/l) (ULN 72) GGT (u/l) (ULN 29) Bilirubin (mg/dl) (ULN 1.10)
Patients randomized to atorvastatin 48/F 440 669 329 179 222 0.72 41/F 234 344 162 116 118 0.89 52/F 291 260 133 227 150 1.21 49/F 194 280 163 72 134 0.62 60/M 273 179 160 68 78 0.83 69/F 137 94 171 441 1088 1.40 66/F 135 162 95 232 152 1.24 55/F 127 197 79 104 74 0.47 37/F 119 96 254 82 95 2.07 59/F 112 110 65 90 122 0.47 69/M 111 95 84 272 579 0.47 58/F 103 195 59 126 174 0.57 61/F 102 142 72 196 151 0.58 53/F 87 120 50 207 192 0.57 37/M 87 89 66 62 18 0.96
Patients randomized to simvastatin 46/M 350 369 176 74 132 0.89 45/F 82 108 58 51 24 0.42
| Pravastatin Reduces Levels of Both Small and Large LDL Particles |